6 Alignement, Read Groups, Mark duplicates
Once the fastq files are ready to be processed we can align them with STAR. Subread/Rsubread is another widely used RNA-Seq aligner. The reason why I initially choose STAR over Subread was simply due to the fact that STAR can generate specific output for chimeric reads that can be directly used with STAR-Fusion to analyse gene fusions (see more in Section 10). Also, STAR is suggested in the the GATK Best Practices to call variants in RNA-Seq.
6.1 Create STAR index
STAR requires an index for the reference genome that will be used for the alignment and fusion calling steps. Below, I show my way of creating the index!
# Iniaitlise Genome directory where to save STAR Index and STAR Fusion Index folders
star_genome100=path_to_genome_directory/star_index_hg19_99
mkdir -p ${star_genome100}To build the STAR index one needs to provide the FASTA file for the reference genome used, a GTF file with information aabout the annotation and STAR also require an extra parameter called sjdbOverhang which is usually set to be (read length - 1). See STAR documentation for Generating genome indexes in the STAR manual
- 99 is (read length - 1) relative to the samples that I was working with.
Below is a wrapper for the STAR call to build an index. You can find the function in the functions folder.
outpud_dir=./results
./functions/build_STAR_index.sh $genome_fasta_path $gtf_path $outpud_dir "hg19" 996.2 STAR-1pass
If you are working with a cohort of bamfiles, the developer of STAR suggests running the alignment in a two-pass mode. This consists in first aligning all the bamfiles with STAR, then collecting the splice junctions output of STAR and finally realigning all the bamfiles with this new information. For more details about STAR 1-pass, 2-pass-multi and 2-pass-single modes see Section 8 of the STAR documentation. In my pipeline I normally use the 2-pass multi mode which consists of a first star 1-pass and a subsequent 2-pass multi run, as shown below.
FQ1=./data/SRR1608907_1.fastq.gz
FQ2=./data/SRR1608907_2.fastq.gz
star_genome100=path_to_genome_directory/star_index_hg19_99
Rscript ./functions/run_STAR.R --genome_index $star_index_hg19_99 \
--fastqfiles $FQ1,$FQ2 \
--sampleName SRR1608907 \
--outdir ./results/star_1pass \
--STARmode "1Pass" The R function above is a wrapper for the STAR call below:
# Version STAR/2.5
STAR --genomeDir path_to_star_index_hg19 \
--readFilesIn $FQ1 $FQ2 --runThreadN 27 --chimSegmentMin 10 --readFilesCommand zcat --alignSJoverhangMin 8 --outBAMcompression 10 --alignSJDBoverhangMin 1 --limitBAMsortRAM 85741557872 --outFilterMismatchNmax 999 --alignIntronMin 20 --alignIntronMax 200000 --alignMatesGapMax 20000 --outFileNamePrefix path_to_star_1pass/SampleName --outSAMtype BAM SortedByCoordinate --outFilterType BySJout --outFilterMultimapNmax 15To see all the arguments available:
Rscript ./functions/run_STAR.R --help
Options:
--genome_index=GENOME_INDEX
Path to the folder with the reference genome index.
--fastqfiles=FASTQFILES
One or two comma separated full paths to the gzipped fastq files.
If only one file is given STAR will consider it a SE library.
--sampleName=CHARACTER
Name for output files. If not specified: --fastqfiles without directory and extention.
--outdir=CHARACTER
Path to output directory. If not specified: ../STAR_align.
--sjfile=CHARACTER
Path to output splije junction file from STAR 1-pass. Required if --STARmode '2PassMulti'.
--STARmode=CHARACTER
One of: '2PassMulti', '2PassBasic', '1Pass'. For more information see the STAR manual for STAR 2-pass mode (Section 8)
--Rrepos=RREPOS
Redirection to server worldwide. Need the default when installing packages without setting a mirror.
--RlibPath=RLIBPATH
R path to install R packages.
-h, --help
Show this help message and exitAfter running STAR on all the fastq files available we can collect all the splice junctions from the first pass and use them for the second pass.
# concatenate splice junctions from all samples from ran in pass1
cat ./results/star_1pass/*SJ.out.tab > ./results/star_1pass/combined_sj.out.tab
# Dobin suggests to remove chrm cause they are usually False positives
awk '!/chrM/' ./results/star_1pass/combined_sj.out.tab > ./results/star_1pass/combined_sj_nochrM.out.tabAgain, for quality check, have a look at the amazing alignment summary enabled by MultiQC.
multiqc ./results/star_1pass --interactive -n "STAR_1passQC" -o ../results6.3 STAR-2pass
The second pass alignment is exactly the same as the first one with only a few differences:
- the sjfile in input. This was created combining the splice junctions from the first pass above.
- STAR is the run with the chimeric reads option switched on, which is necessary if you need to analyse fusion genes.
The ouput of STAR will be a bamfile already sorted by coordinate with the suffix Aligned.sortedByCoord.out.bam.
FQ1=./data/SRR1608907_1.fastq.gz
FQ2=./data/SRR1608907_2.fastq.gz
star_genome100=path_to_genome_directory/star_index_hg19_99
Rscript ./functions/run_STAR.R \
--genome_index $star_index_hg19_99 \
--fastqfiles $FQ1,$FQ2 \
--sampleName SRR1608907 \
--outdir ./results/star_2pass --STARmode "2PassMulti" \
--sjfile ./results/star_1pass/combined_sj_nochrM.out.tabAt this stage we can also run two more steps post_align_qc1.sh and post_align_qc2.sh, discussed below.
# Run featurecounts and collect fragment sizes for QC
./functions/post_align_qc1.sh \
path_to_genome.fa \
path_to_genes.gtf \
./results/star_2pass/SRR1608907.Aligned.sortedByCoord.out.bam \
SRR1608907 # sample name
# Pre-process bamfile (add Read groups etc..)
./functions/post_align_qc2.sh \ ./results/star_2pass/SRR1608907.Aligned.sortedByCoord.out.bam \
SRR1608907 \
path_to_genome.fa \
SRR1608907 6.4 Details about post-alignment functions
post_align_qc1.shis optional:- Runs featureCounts to get gene counts and compute PCA to evaulate the concordance between bamfiles sequenced on different lanes. This allows a QC before merging the different bamfiles into a single one.
- Runs CollectMultipleMetrics to collect the fragment distribution of the bamfiles (only possible with PE reads). This is also a good QC to check that the fragment distribution of bamfiles on different lanes is the same.
post_align_qc2.shcontains necessary pre-prcessing steps:- Marks PCR duplicates (using sambamba markdup)
- Add Read Groups to single runs before merging bamfiles (using AddOrReplaceReadGroups). Even if files do not need to be merges across lanes,
GATKrequires read groups to be added to bamfiles. - Run ValidateSamFile to check for errors in the final bamfile.
In order, post_align_qc2.sh arguments are:
- Path to aligned bamfile;
SampleName. This is the name of the sample applied to theRGIDandRGPUfields below.SampleName of the run. If a sample was sequenced across different lanes you need to set lane-specific read groups to each separate bamfile (e.g. SampleName_L1, SampleName_L2). This sample name will be used for the fieldsRGLBandRGSMin theAddOrReplaceReadGroupsgroups below. See Section 6.5 for merging bamfiles.
# Picard tool function to add read groups to a bamfile
AddOrReplaceReadGroups \
I= ./star_2pass/SRR1608907.Aligned.sortedByCoord.out.bam \
O= ./star_2pass/SRR1608907.Aligned.sortedByCoord.out.RG.bam \
RGID=SRR1608907 \
RGPU=SRR1608907 \
RGLB=SRR1608907_L1 \
RGPL="illumina" \
RGSM=SRR1608907_L1Figure 6.1 is a graphic example of adding read groups to separate files. In the merged bamfile each reads will have an identification for which lane they came from and this information is used in the GATK Base Recalibration step discussed in Section 7.
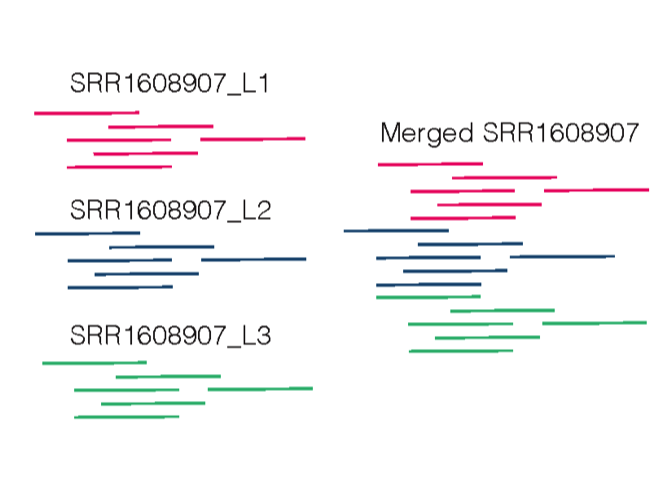
Figure 6.1: Adding read group before merging across lanes.
After running post_align_qc2.sh a file with the suffix Aligned.reorderedDupl.rg.bam will be created where read groups are added and PCR duplicated reads marked.
This time MultiQC will give us a summary output also of the fragment distributions and gene counts if all the output files are stored within the star_2pass folder.
multiqc ./results/star_2pass --interactive -n "STAR_2passQC" -o ../results6.5 Merge bamfiles
In some cases the sequenced reads from one sample can be sequenced across different lanes and the aligned bamfiles need to be merged. sambamba merge can be used for this. I created a wrapper function for it even though it assumes that the files from the same sample have a common SampleName. The function will merge together all bamfiles containing SampleName found in the ./star_2pass directory.
./functions/merge_runs.sh SampleName ./star_2pass