From FASTQ files to Variant Calling for RNA-Seq
1 Setup
This is an example workflow from SRR files to Variant calling using modular functions written in R and bash.
git clone git@github.com:annaquaglieri16/RNA-seq-variant-calling.git
cd ./RNA-seq-variant-callingAll the functions used for the variant calling and downsampling pipeline are inside the ./functions folder.
- If you want to download sample
FASTQfiles or learn how to downloadFASTQfiles from GEO go to Section 2. - If you already have the
FASTQfiles and YOU WANT TO randomly downsample your samples to a fix number of reads go to Section 3. - If you already have the
FASTQfiles and you don’t need to perform quality control or downsampl your files go to Section 6. - If you already have the
BAMfiles and you want to call variants go to Section 7.
1.1 Overview
Figure 1.1 below offers an overview of the pipeline that I applied to several of the cancer RNA-Seq samples that I worked with. However, the current book mentions other callers not displayed in the figure.

Figure 1.1: Overview of the variant calling pipeline that I used used for several cancer RNA-Seq data.
The sofwtare mentioned in Figure 1.1 are mentioned throughout the book and cited below:
GATK(McKenna et al. 2010)VarScan(Koboldt et al. 2012)superFreq(Flensburg et al. 2018)VarDict(Lai et al. 2016)km(Eric Olivier Audemard, Patrick Gendron, Vincent-Philippe Lavallée, Josée Hébert, Guy Sauvageau, Sébastien Lemieux 2018)VEP(McLaren et al. 2016)varikondo
The pre-processing steps in Figure 1.1 are also summarised in Figure 1.2 and discussed in the sections below. The majority of the pre-processing steps are taken from the GATK best practices for RNA-Seq variant calling.
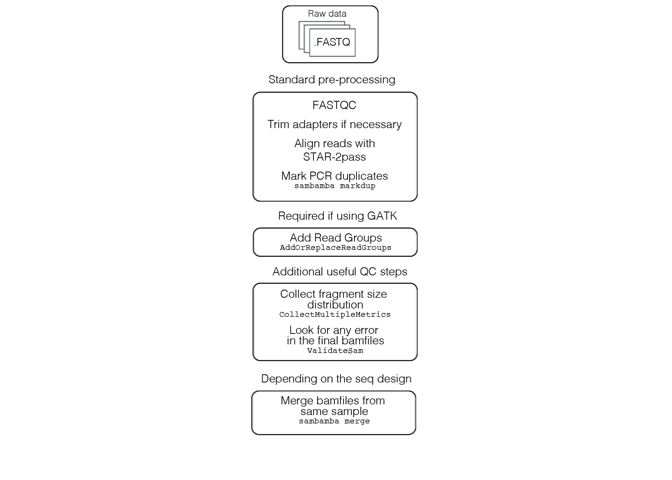
Figure 1.2: Bamfile pre-processing.
1.2 Disclaimer
The following workflow was built in a modular way and it is not wrapped up into a pipeline manager. I aknowledge the limitations and non-user-friendliness of some steps. However, it offers a comprehensive view of several tools and steps used for variant calling in RNA-Seq as well as general tools used in any bioinformatics pipeline.